Mikko Taipale
PhD

Qualification
- Whitehead Institute, Cambridge, MA, U.S., Research fellow in Susan Lindquist’s laboratory, 2007-2014.
- European Molecular Biology Laboratory, Heidelberg, Germany, PhD in Molecular Biology (with Asifa Akhtar), 2001-2005.
- University of Oulu, Finland, MSc in Genetics, 1995-2000.
MY RESEARCH OVERVIEW (GO TO SCIENTIFIC OVERVIEW)
Functional proteomics and protein homeostasis
Research in my laboratory can be loosely divided into three areas: basic research, technology development, and translational research.
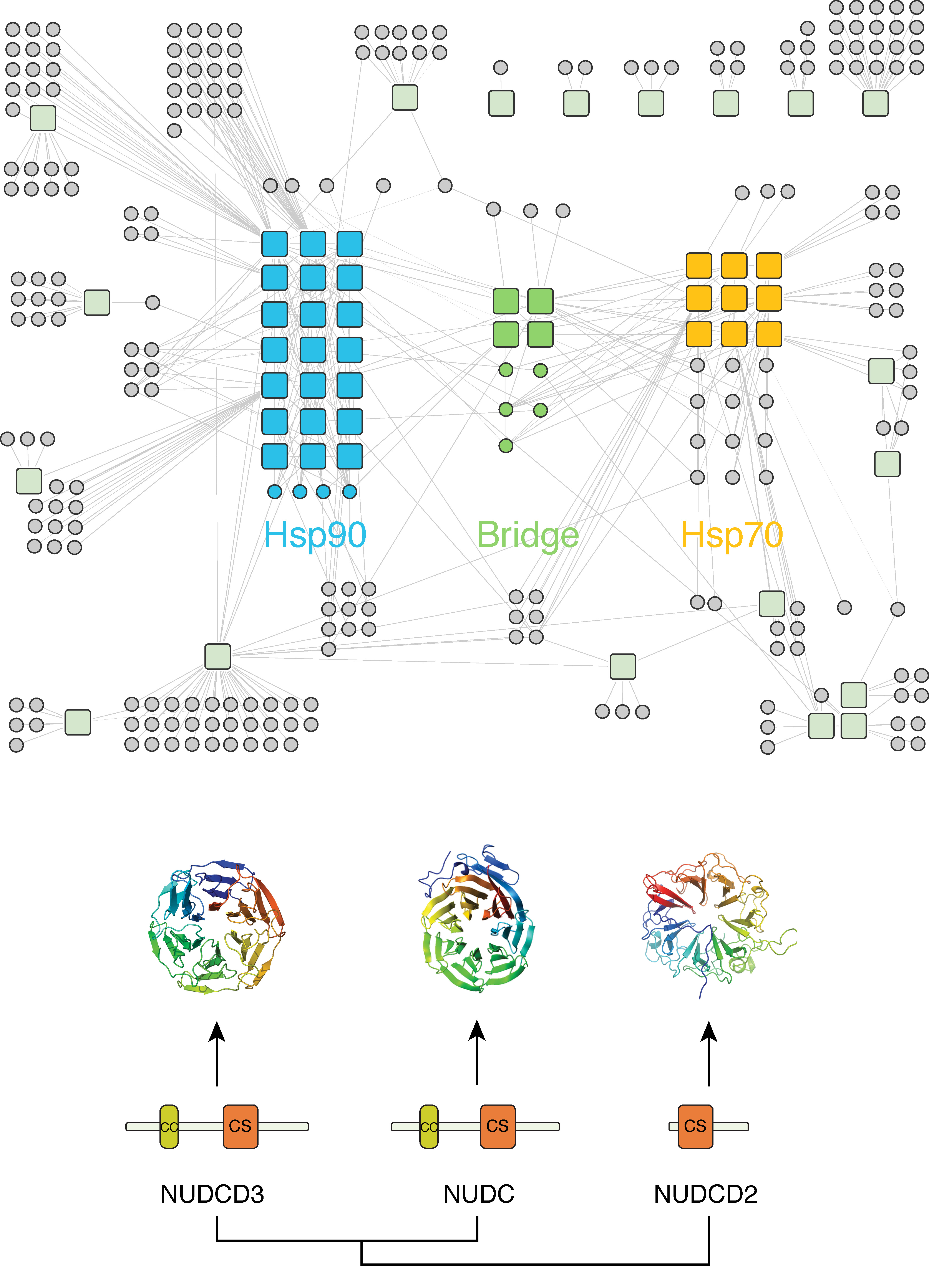
From the basic research perspective, we are interested in the life cycle of proteins. How do they adopt the right three-dimensional structure in a crowded environment? How do they find the right location in the cell? And how are they degraded when they are not needed any more or when they do not function properly? We employ various large-scale protein/protein interaction assays to to understand the organization of the machinery that regulates these processes. But the network itself is not the goal. Rather, we use large-scale data to ask specific questions. Why has the eukaryotic chaperone machinery evolved such a large cohort of regulatory factors and co-chaperones? How are these cofactors regulating chaperone function? How do chaperone/client interaction networks change in response to cellular conditions, or during evolution? Why do proteins need chaperones in the first place? We are also interested in scaffold proteins and other hubs that, like chaperones, interact with hundreds of different client proteins and regulate diverse cellular processes.
The second area of our research is developing tools for detecting and characterizing biomolecular interactions. In particular, we are interested in interactions that have been traditionally difficult to characterize in a systematic manner. These include e.g. ligand/receptor interactions and E3 ligase/target protein interactions. Furthermore, we are taking advantage of our large chaperone/client interaction networks to develop thermodynamic and conformational sensors for drug/target interactions. These assays can be used to study target engagement of small molecules in living cells and to discover novel targets for clinically used compounds.
The third part of our research is more translational. In collaboration with Marc Vidal’s laboratory in the Dana-Farber Center for Cancer Systems Biology, we have generated a collection of over 2,500 mutant alleles for 1,100 rare Mendelian disorders. This collection facilitates analysis of disease-causing mutations in a systematic way. We will functionally characterize these mutations in various cellular assays and investigate the fundamental molecular mechanisms that cause rare diseases. We will also characterize the effects of known small-molecule drugs on these mutant proteins. Essentially, our aim is to find a disease for a drug, not a drug for a disease.
SCIENTIFIC RESEARCH OVERVIEW
Current projects in our lab are organized along the three streams:
1. Protein homeostasis in human cells
It’s tough to be a protein. From the moment of birth in the ribosomal exit tunnel until the very last squeeze through the proteasomal channel, they are often harassed by various stresses like heat, ionizing radiation, and reactive oxygen species. To cope with this, cells have evolved an elaborate network of proteins that maintain protein homeostasis, or proteostasis. This evolutionary ancient network consists of chaperones and co-chaperones, E3 ligases and deubiquitinases, trafficking proteins, and hundreds of other quality-control factors. In turn, these factors associate with and regulate essentially all cellular proteins.
We are interested in deciphering how this network is physically organized in human cells. We use diverse high-throughput protein/protein interaction assays to characterize the network and identify the key players. However, the network itself is not the goal. Rather, we use the large-scale data to ask specific questions. Why has the eukaryotic chaperone machinery evolved such a large cohort of regulatory factors and co-chaperones? How are these cofactors regulating chaperone function? How do chaperone/client interaction networks change in response to cellular conditions, or during evolution? Why do proteins need chaperones in the first place?
2. Rare diseases
There are about 7,000 known rare diseases. While each of them is rare, together they affect millions of people around the world. In the next 5 years, we will essentially know all the genes underlying Mendelian diseases, which account for most rare diseases. But what then? There is no cure nor treatment for most of these diseases, and pharmaceutical companies have no financial incentives to develop drugs for them.
The traditional approach has been to use multiple assays and techologies to study a particular Mendelian disease. But what if we turned the question upside down? What if we studied all rare diseases at the same time but with very specific assays. For example, instead of screening thousands of different drugs to treat one disease, could we screen thousands of diseases with one drug?
As a collaboration between Marc Vidal’s lab at the Center for Cancer Systems Biology and Sue Lindquist’s lab at the Whitehead Institute, we have created a Gateway-compatible collection of almost 3,000 mutant alleles for 1,100 Mendelian diseases (Sahni, Song, Taipale et al. Cell 2015). Our aim is to systematically phenotype this collection of disease-causing alleles in order to find novel connections between poorly-studied rare diseases and known pathways.
3. Technology development
We believe that technology drives innovation as much as the other way around. On the one hand, restriction enzymes, RNAi, reprogramming, and CRISPR are all fascinating biological phenomena in their own right, but this is dwarfed by their impact as enabling technologies. On the other, although technologies such as next-gen sequencing were originally developed with specific applications in mind, researchers have exploited and repurposed the platforms for their own research.
Our aim is to develop methods and tools to investigate biomolecular interactions that have not been amenable to traditional methods. These include interactions between E3 ligases and their targets; secreted ligands and their receptors; and small molecule drugs and their cellular targets. For example, we have shown that chaperones can be exploited as “thermodynamic sensors” to detect drug/target interactions in living cells (Taipale et al. 2013). Systematic characterization of the chaperone/client interaction network enables us to expand the scope of this method to novel drug target classes.
Although we are certainly interested in fundamental biological questions, our aim here is to let the results guide us and take us to new research avenues.
SELECT PUBLICATIONS
- Widespread macromolecular interaction perturbations in human genetic disorders. Sahni N, Yi S, Taipale M, et al. Cell. 2015 Apr 23;161(3):647-60.
- A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, Choi H, Berger B, Gingras AC, Lindquist S. Cell. 2014 Jul 17;158(2):434-48.
- Chaperones as thermodynamic sensors of drug-target interactions reveal kinase inhibitor specificities in living cells. Taipale M, Krykbaeva I, Whitesell L, Santagata S, Zhang J, Liu Q, Gray NS, Lindquist S. Nat Biotechnol. 2013 Jul;31(7):630-7.